Genomic analysis I (KCSPK 21) - Identified spontaneous mutations or induced mutations
in genomes by chemical or physical agents.
This chapter will examine the technologies used in Genome
- Generated linkage maps using these mutant strains.
Analysis and the data generated for di9erent species.
- Identified genes in model organisms like Drosophila,
Accompanying technologies comprising the broader ‘Omics’, like
mice, maize, yeast, bacteria and viruses.
Transcriptomics, Proteomics and Bioinformatics will also be
- Many disadvantages: require a mutation in a gene
introduced.
before a linkage map can be constructed; very slow;
The chapter will be presented in six lectures: only in non-human organisms.
• In the 1980s, recombinant DNA technology was used to
1. Earlier genomic analysis approaches; Whole genome
map human DNA sequences to specific chromosomes.
sequencing
- These sequences were not full-length genes but marker
2. Bioinformatic applications and Genome databases
sequences such as restriction fragment length
3. Functional genomics and the Human Genome Project
polymorphisms (RFLPs).
4. Omics
- Once these markers were assigned to chromosomes, it
5. Comparative genomics; Metagenomics
could be used to establish linkages between the
6. Transcriptome analysis; Proteomics
markers and disease phenotypes for genetic disorders.
Earlier genomic analysis approaches - Allowed for more than 3500 genes and markers to be
Genomics before modern sequencing technology mapped to human chromosomes.
• The genomics era was introduced by Sanger et al. when • In the 1990s, human genes estimated at ±100 000 –
they sequenced the 5400 bp genome of the phage ɸX174 in impossible to map and clone using traditional methods
1977 Whole genome sequencing
• Sequencing technology have exploded so that we now Genomics allows sequencing of entire genomes
experience a Genomics revolution
• Most widely used strategy for sequencing and assembling
• Quickly followed by transcriptomics, proteomics,
an entire genome involves variations of a method called
metabolomics [Omics] … AND Bioinformatics
whole-genome sequencing (WGS) or shotgun cloning.
Earlier genomic analysis approaches • Restriction digests (or sonication) of whole chromosomes
• Geneticists typically followed a two-part classical genetics generate thousands to millions of overlapping DNA
approach to identify and characterize the genes in an fragments.
organism’s genome: • Sequenced and assembled using Bioinformatic
applications.
, - Software that creates DNA sequence alignments. Continuous fragments (contigs)
- Alignments identify overlapping sequences, which can
be used to map onto chromosomes.
- Overlapping sequences are adjoining that together form
a continuous DNA fragment, called a contig.
• The WGS shotgun method was developed by J. Craig Venter
at The Institute for Genome Research (TIGR) in 1995, when
they sequenced the 1.83-million-bp genome of the
bacterium Haemophilus influenzae.
• This was the first complete genome sequence from a free-
living (i.e. nonviral) organism demonstrating “proof-of-
concept” that shotgun sequencing could be used to
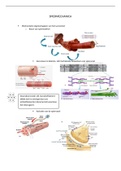
• In the example above, alignment software has identified an
sequence an entire genome.
overlap between three fragments of sequenced DNA
(contigs 1, 2 and 3) from human chromosome 2. The
software can assemble the three sequences into one much
larger sequence using the overlaps. In this way, the
sequence of the entire chromosome can be assembled in
silico
High-throughput Sequencing (HTS)
Conventional sequencing is too slow for WGS
• Computer-automated DNA sequencers
- Designed for high-throughput sequencing, thus making
genomics possible
- Essential for Human Genome Project
- Sequencers contained multiple capillary gels (96)
- Generated over 2 million bp per day
• Sequencing cost and time have decreased remarkably
- The major technological breakthrough that
revolutionized genomics possible was the advent of
, Next-Gen Sequencing (NGS), like the 454-
pyrosequencing method.
- Sequencing the first human genome cost about $1
billion and took 13 years to complete; today it costs <
$1000 and takes a day
- Moore’s law
Compiling genome sequences
WGS: the clone-by-clone approach
Draft sequences and reference genomes
a.k.a map-based cloning
• First assemblies generate draft genomes.
• DNA fragments from restriction digests are aligned,
• Final assemblies constitute reference genomes.
creating a restriction map of the chromosome.
• A reference genome is never 100% final - “final” is dictated
• Restriction fragments are then ligated into vectors such as
by the number of errors that are accepted as a cut-o9.
BACs or YACs to create libraries of contigs.
• Accuracy improved by sequencing multiple times.
• These can be further digested into smaller, more easily
• Coverage (or depth) is the number of times that a particular
manipulated pieces that are subcloned into smaller
nucleotide appears in the same position after multiple
vectors such as plasmids.
reads have been compiled.
• Contigs are sequenced and aligned to assemble the entire
• Once compiled/error checked, the genome is analyzed to
chromosome.
identify:
• Initial progress on the Human Genome Project was based
- Gene sequences
on this methodology.
in genomes by chemical or physical agents.
This chapter will examine the technologies used in Genome
- Generated linkage maps using these mutant strains.
Analysis and the data generated for di9erent species.
- Identified genes in model organisms like Drosophila,
Accompanying technologies comprising the broader ‘Omics’, like
mice, maize, yeast, bacteria and viruses.
Transcriptomics, Proteomics and Bioinformatics will also be
- Many disadvantages: require a mutation in a gene
introduced.
before a linkage map can be constructed; very slow;
The chapter will be presented in six lectures: only in non-human organisms.
• In the 1980s, recombinant DNA technology was used to
1. Earlier genomic analysis approaches; Whole genome
map human DNA sequences to specific chromosomes.
sequencing
- These sequences were not full-length genes but marker
2. Bioinformatic applications and Genome databases
sequences such as restriction fragment length
3. Functional genomics and the Human Genome Project
polymorphisms (RFLPs).
4. Omics
- Once these markers were assigned to chromosomes, it
5. Comparative genomics; Metagenomics
could be used to establish linkages between the
6. Transcriptome analysis; Proteomics
markers and disease phenotypes for genetic disorders.
Earlier genomic analysis approaches - Allowed for more than 3500 genes and markers to be
Genomics before modern sequencing technology mapped to human chromosomes.
• The genomics era was introduced by Sanger et al. when • In the 1990s, human genes estimated at ±100 000 –
they sequenced the 5400 bp genome of the phage ɸX174 in impossible to map and clone using traditional methods
1977 Whole genome sequencing
• Sequencing technology have exploded so that we now Genomics allows sequencing of entire genomes
experience a Genomics revolution
• Most widely used strategy for sequencing and assembling
• Quickly followed by transcriptomics, proteomics,
an entire genome involves variations of a method called
metabolomics [Omics] … AND Bioinformatics
whole-genome sequencing (WGS) or shotgun cloning.
Earlier genomic analysis approaches • Restriction digests (or sonication) of whole chromosomes
• Geneticists typically followed a two-part classical genetics generate thousands to millions of overlapping DNA
approach to identify and characterize the genes in an fragments.
organism’s genome: • Sequenced and assembled using Bioinformatic
applications.
, - Software that creates DNA sequence alignments. Continuous fragments (contigs)
- Alignments identify overlapping sequences, which can
be used to map onto chromosomes.
- Overlapping sequences are adjoining that together form
a continuous DNA fragment, called a contig.
• The WGS shotgun method was developed by J. Craig Venter
at The Institute for Genome Research (TIGR) in 1995, when
they sequenced the 1.83-million-bp genome of the
bacterium Haemophilus influenzae.
• This was the first complete genome sequence from a free-
living (i.e. nonviral) organism demonstrating “proof-of-
concept” that shotgun sequencing could be used to
• In the example above, alignment software has identified an
sequence an entire genome.
overlap between three fragments of sequenced DNA
(contigs 1, 2 and 3) from human chromosome 2. The
software can assemble the three sequences into one much
larger sequence using the overlaps. In this way, the
sequence of the entire chromosome can be assembled in
silico
High-throughput Sequencing (HTS)
Conventional sequencing is too slow for WGS
• Computer-automated DNA sequencers
- Designed for high-throughput sequencing, thus making
genomics possible
- Essential for Human Genome Project
- Sequencers contained multiple capillary gels (96)
- Generated over 2 million bp per day
• Sequencing cost and time have decreased remarkably
- The major technological breakthrough that
revolutionized genomics possible was the advent of
, Next-Gen Sequencing (NGS), like the 454-
pyrosequencing method.
- Sequencing the first human genome cost about $1
billion and took 13 years to complete; today it costs <
$1000 and takes a day
- Moore’s law
Compiling genome sequences
WGS: the clone-by-clone approach
Draft sequences and reference genomes
a.k.a map-based cloning
• First assemblies generate draft genomes.
• DNA fragments from restriction digests are aligned,
• Final assemblies constitute reference genomes.
creating a restriction map of the chromosome.
• A reference genome is never 100% final - “final” is dictated
• Restriction fragments are then ligated into vectors such as
by the number of errors that are accepted as a cut-o9.
BACs or YACs to create libraries of contigs.
• Accuracy improved by sequencing multiple times.
• These can be further digested into smaller, more easily
• Coverage (or depth) is the number of times that a particular
manipulated pieces that are subcloned into smaller
nucleotide appears in the same position after multiple
vectors such as plasmids.
reads have been compiled.
• Contigs are sequenced and aligned to assemble the entire
• Once compiled/error checked, the genome is analyzed to
chromosome.
identify:
• Initial progress on the Human Genome Project was based
- Gene sequences
on this methodology.